
RESEARCH ARTICLE
Development, Characterization and Transdermal Delivery of Dapsone and an Antibiotic Entrapped in Ethanolic Liposomal Gel for the Treatment of Lapromatous Leprosy
Ruchi Tiwari*, Gaurav Tiwari, Pranay Wal, Ankita Wal, Priyanka Maurya
Article Information

Identifiers and Pagination:
Year: 2018Volume: 5
First Page: 1
Last Page: 15
Publisher Id: TONMJ-5-1
DOI: 10.2174/1875933501805010001
Article History:
Received Date: 30/11/2017Revision Received Date: 5/4/2018
Acceptance Date: 16/4/2018
Electronic publication date: 30/04/2018
Collection year: 2018
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: https://creativecommons.org/licenses/by/4.0/legalcode. This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Background and Objective:
Applying Ethosomal Gels (EGs) in transdermal drug delivery systems has evoked considerable interest because of their good water-solubility and biocompatibility. The aim of present study was to prepare and characterize ethosomes of antileprotic drug Dapsone (DAP) together with an antibiotic Cloxacillin Sodium (CLXS) which may deliver these drugs to targeted site more efficiently than marketed gel preparation of DAP and also overcome the problems related with oral administration of CLXS.
Methodology:
Ethosomes were prepared by cold method then characterized for particle size, Entrapment Efficiency (EE), zeta potential and permeation studies. Vesicular size was determined by Scanning Electron Microscopy (SEM) and found to be varied from 127±9.01 to 215±7.23 nm depending on the concentrations of soya lecithin and ethanol.
Results:
The average percent drug entrapment efficiency of formulations ranged between 52.31% to 73.51% and 49.07% to 71.91% for DAP and CLXS respectively. The high ethanol concentration in ethosomes has shifted the vesicular charge from positive to negative. It was observed that F1 and F2 formulations were having zeta potential of -25.08±1.03 mV and -50.11±1.97 mV respectively and do not aggregate rapidly. The drug release of ethosomes ranged from 84.68% to 96.58% and 64.89% to 84.21% for DAP and CLXS respectively. Ethosomal gel was prepared with optimized ethosome and studied for its release and physicochemical characteristics.
Conclusion:
Finally, G5 demonstrated better (p < 0.05) antileprotic effect to improve effectiveness, stability and to reduce side effects and toxicity associated with the chosen drugs in order to treat Leprosy.
1. INTRODUCTION
Leprosy, also known as Hansen’s disease, is a worldwide health problem, varying from tuberculoid to Lepromatous Leprosy (i.e., paucibacillary to multibacillary disease) according to the host immune response. It is caused by Mycobacterium leprae, and affects the skin, eyes, and nerves, leading to skin lesions, eye pain, loss of vision, weakness, and numbness. The final diagnosis is based on a combination of findings on skin biopsy, smear, and physical examination. The treatment options differ according to the clinical manifestations. Leprosy can be associated with type 1 (reversal) and type 2 (erythema nodosum leprosum) immunologic reactions that may occur at any time before, during, or after the start of treatment. During this process, this disease can worsen dramatically, and antibacterial therapy should continue during these periods [1]. DAP (4,4′-diaminodiphenylsulfone) is a drug of the sulfone class and was discovered in 1908. A unique property of DAP is that it has dual therapeutic activity and demonstrates antimicrobial and anti-inflammatory properties. Since 1943, it has been the drug of choice in the treatment of leprosy because it has bacteriostatic action against Mycobacterium leprae by inhibiting folic acid production via competition with p-aminobenzoate for the active site of dihydropteroate synthase [2]. It has obvious application in the field of dermatology, and may ease symptoms in diseases such as acne, Behçet’s disease, epidermolysis the bulbous acquisita, dermatitis herpetiformis, Kaposi’s sarcoma, and systemic lupus erythematosus [3]. DAP is classified as a class II drug according to the Biopharmaceutics Classification System, and has high permeability and low solubility in water (log P = 0.97). Thus, despite its therapeutic potential, the low solubility of DAP in water results in low bioavailability and microbial resistance. Oral administration of DAP is associated with several adverse effects, including hemolytic anemia, peripheral neuropathy, nausea, and headache. These side effects diminish its feasibility for treating skin diseases by the oral route. Many of the adverse effects of DAP are related to the production of metabolites. In the liver, DAP is acetylated by N-acetyltransferase which produces monoacetyl DAP, and upon enzymatic hydroxylation, DAP hydroxylamine is produced, which is primarily responsible for the development of adverse effects [4, 5]. Because of the therapeutic relevance of DAP, it is desirable to reduce its adverse effects using nanotechnology [6]. Most infections due to Gram-positive organisms can be treated with quite a small number of antibiotics. Penicillin, CLXS, and erythromycin should be enough to cover 90 percent of Gram-positive infections. The relatively narrow spectrum of these drugs should be the incentive to prescribers to use them selectively, together with adequate bacteriological investigation, in order to achieve effective treatment with a minimum of disturbance to the patient's normal bacterial flora and without any other harmful side effects [6, 7]. CLXS, chemically monosodium (2S, 5R, 6R)-6-{[3-(2-chlorophenyl)-5-methyloxazole-4-carbonyl] amino}-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo [3.2.0] heptane-2- carboxylic acid is a semisynthetic antibiotic in the same class as penicillin. It is the chlorinated derivative of ofloxacin and ofloxacin is used in the treatment of leprosy as an antibiotic drug. It is prescribed for the treatment of a wide range of bacterial infections caused by susceptible organisms including soft-tissue infections such as boils or infections as a result of spider bites, ear infections, pneumonia, impetigo - a bacterial skin infection characterised by small pus-filled blisters, and endocarditis - inflammation of the lining of the heart and its valves [8]. It works by binding to specific Penicillin Binding Proteins (PBPs) located inside the bacterial cell wall and inhibiting the third and last stage of bacterial cell wall synthesis. Cell lysis is then mediated by bacterial cell wall autolytic enzymes such as autolysins, it is possible that CLXS interferes with an autolysin inhibitor. Utilization of topical applications provides an efficient strategy both for the alternative treatment of leprosy, where the pathogen and lesions are in the skin, and other dermatological indications, such as acne. Among the various colloidal nanosystems available, ethanol vesicles are notable because they offer recognized advantages by increasing the solubility of hydrophobic drugs such as DAP. Ethosome carriers were first reported by Touitou et al. and are a modified form of liposome. Ethosomes are vesicle lipid carriers containing phospholipid, and have a high concentration of ethanol and water. The ethosome has an ability to efficiently entrap various molecules, like hydrophilic, lipophilic and amphiphilic molecules. It can be explained by the high degree of lamellarity and the presence of ethanol in the vesicles, which allows for better solubility of many drugs. Moreover, ethosomal formulations possess greater entrapment capability than liposome [9]. One of the important characteristics of ethosomes is due to their soft and flexible nature, they can penetrate the skin and allow enhanced delivery of various active agents to deeper strata of the skin or enhanced systemic circulation compared to conventional liposome or hydroethanolic solutions [10]. Better permeability of ethosome carriers may be due to the synergistic mechanism between high concentration of ethanol, phospholipids vesicles and skin lipids. Many studies have used ethosome as a carrier system of drugs delivery for the transdermal or topical administration. It has been reported to improve various drug deliveries both in vivo and in vitro skin, such as Testosterone, Cannabidiol, Buspirone hydrochloride, Erythromycin, Ammonium glycyrrhizinate, Lamivudine and 5-aminolevulinic acid. In recent times, ethosomes have attracted considerable attention for their opportunity to enhance dermal/transdermal delivery of a number of bioactive agents [11].
The main aim of the study is to attain effective drug concentration at the intended site of action for a sufficient period of time to elicit the response. Ethosomal gel had been prepared with CLXS and DAP using carbopol 934 as gelling polymer to improve effectiveness, stability, bioavailability and to reduce side effects and toxicity associated with the chosen drugs in order to treat Leprosy.
2. MATERIALS AND METHODS
DAP (DAP) and Cloxacillin Sodium (CLXS) were kindly provided by Yarrow chem. products, Mumbai, India and Penam laboratories Ltd., Dharuhera (Haryana) respectively. Cholesterol and Soy Lecithin were purchased from Central drug house, New Delhi. Propylene Glycol, Carbopol 934 and PEG 400 were purchased from SD Fine Chemical Limited, Mumbai, India. Cellophane membrane (molecular weight cut off 12,000-14,000) was purchased from Qualigens Fine Chemicals, Mumbai, India. Ethanol and all other chemicals used were of analytical grade.
2.1. Phase Solubility Studies
In the phase solubility study, an excess amount of different drugs (10 mg) was added individually into 10 ml of distilled water containing various concentrations of cholesterol ranging from 0.2-0.8% w/v and shaken for 72 hrs in a water bath shaker at 250C±10C with 100 rpm. Samples were filtered through a bold>nylon filter disk (0.22 μ) and assayed against the blank prepared by using different concentrations of cholesterol in distilled water [12].
2.2. In Vitro Adsorption Studies
Ten milligrams of different drugs were added individually to 50 ml of distilled water and tenfold weight (100 mg) of soya lecithin was added to it. The mixture was magnetically stirred at room temperature (25°C) for 24 h. Samples (5 ml) were periodically drawn at 0, 1, 2, 3, 4, 5, 6, 7, 8, 18, 20, 22 and 24 h and assayed spectrophotometrically at 293 and 271 nm using UV Spectrophotometer (Shimadzu, Pharma Spec 1700, Kyoto, Japan) for DAP and CLXS respectively to determine the percent drug adsorbed with respect to time [12].
2.3. Quality Target Product Profile (QTPP) and Risk Analysis of DAP and CLXS
The QTPP is described as the quality properties that a drug product need to possess so as to fulfill the objectives set in target product profile as quantitative attributes. QTPP should furnish a quantitative surrogate to describe the aspects of clinical safety and efficacy. A prospective summary of the quality characteristics of a drug product that ideally will achieve to ensure the desired quality, taking into account safety and efficacy of the drug product [13].
2.4. Preparation of Blank and Drug Loaded Ethosomes
Ethosomes were prepared by cold method with certain modifications. Phospholipid (3-5% w/v) and cholesterol (0.5-1% v/v) were dissolved in ethanol (30-40% v/v) at room temperature by vigorous stirring with the use of a mixer. Propylene glycol (5-10% v/v) was added during stirring and the temperature was maintained at 300c in a water bath. Distilled water or drug solution in distilled water (1% w/v solution), which is previously heated up to 30± 1°C, was added slowly in a fine stream of the above ethanolic lipid solution with continuous mixing using a magnetic stirrer at 900 rpm (Fig. 1). Mixing was continued for another 5 minutes and finally, the vesicular dispersions resulted was left to cool at room temperature (25± 1°C) for 45 minutes. Different ethosomal dispersions and their composition are shown in Table 1 [14].
| S.No. | Formulation code | Independent variable |
Zeta Potential (mV) |
Vesicle size (nm) | Entrapment efficiency (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| X1(%)* | X2(%)# | X3(%)$ | X4(%)+ | DAP | CLXS | ||||
| 1. | F1 | 40 | 5 | 1 | 5 | -25.08±1.03 | 127±9.01 | 63.32 | 70.56 |
| 2. | F2 | 40 | 7.2 | 0.48 | 4.4 | -50.11±1.97 | 130±10.20 | 73.51 | 71.91 |
| 3. | F3 | 40 | 10 | 1 | 5.2 | -28.30±2.67 | 138±5.98 | 61.42 | 68.85 |
| 4. | F4 | 30 | 7 | 1.3 | 3.5 | -30.42±1.02 | 211±2.67 | 59.09 | 58.98 |
| 5. | F5 | 36 | 7 | 0.55 | 3.5 | -39.56±2.04 | 198±4.35 | 52.31 | 49.07 |
| 6. | F6 | 40 | 5 | 1 | 5 | -34.71±2.40 | 144±5.87 | 58.98 | 52.19 |
| 7. | F7 | 40 | 10 | 1 | 3 | -25.08±1.03 | 160±6.34 | 55.62 | 61.43 |
| 8. | F8 | 36 | 11 | 0.6 | 3.5 | -29.32±1.41 | 215±7.23 | 73.13 | 65.29 |
| 9. | F9 | 40 | 7 | 1 | 5 | -35.21±2.33 | 141±3.09 | 69.25 | 70.06 |
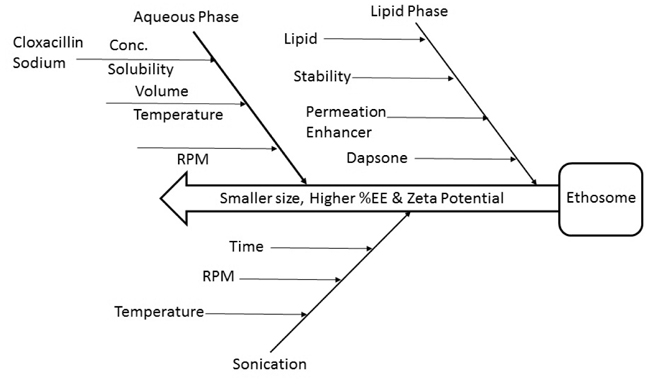 |
Fig. (1). Quality target product profile of ethosome. |
2.5. Effect of Probe Sonication on Optimized DAP and CLXS
It is often sonication which brings about further reduction in size. Sonication breaks coarse drops into nano droplets and hence, it is responsible for obtaining a smaller particle size of ethosome. Sonication effects on the size, Drug content, Drug entrapment, Zeta potential, pH and Drug release (Fig. 2).
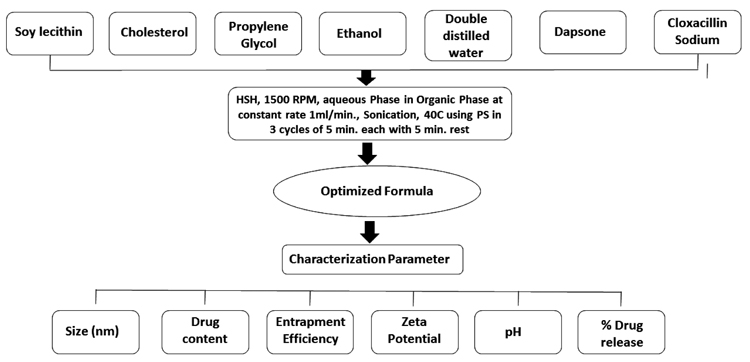 |
Fig. (2). Effect of sonication on optimised ethosome. |
2.6. Characterization of Prepared Ethosomes
To identify the presence of organic functional groups in the prepared ethosomal formulations, FTIR study was performed. The IR Spectra of all prepared formulations were obtained with FTIR Spectrophotometer, Perkin Elmer Spectrum. The scanning range was 4000-400cm-1 at a scan period of 1 minute [15]. Surface morphology of optimized ethosome was observed by Scanning Electron Microscope (SEM). Optimized ethosome was mounted on a glass stub, air-dried and coated with gold using a sputter coater (JEOL, Japan) and finally, visualized under a JEOL JSM - 6490LA scanning electron microscope (JEOL, Japan) at an accelerating voltage of 15 kV from Babasaheb Bhimrao Ambedkar University, Lucknow [16].
2.7. Particle Size Distribution and Zeta Potential Analyzer
The particle size, size distribution and zeta potential of ethosomes was determined by Zeta Potential Analyzer model Zeta PALS (Brookhaven Instruments Co., NY, USA) at 25°C with a scattering angle(s) of 90 degrees after diluted of 10 PL ethosome with 4 mL Milli-Q water. All determinations were performed in triplicate.
2.8. Percent Drug Entrapment Study
Purified vesicular suspension was transferred into a centrifuge tube and centrifuged (R-4°C, Remi centrifuge, Vasai, India) for 1 h at 4,000 rpm. Sediment was lysed using methanol and filtered through filter paper. The drug was assayed both in the sediment and supernatant to determine the entrapment efficiency by the following equation [17].
% Entrapment Efficiency = CL/CI ×100
Where, CL = Amount of drug in sediment, CI = Amount of drug added
2.9. Determination and Evaluation of in vitro Drug Release
In vitro drug release study was performed by using a Franz Diffusion cell. The synthetic cellophane membrane was mounted between the donor and receptor compartment of the diffusion cell. 1 gm of prepared ethosomes were placed over the drug release membrane and the receptor compartment of the diffusion cell was filled with phosphate buffer 7.4. The whole assembly was fixed on a magnetic stirrer and the solution in the receptor compartment was continuously stirred using a magnetic bead at 50 rpm; the temperature was maintained at 37°C. 1 ml sample was withdrawn at specific time points over a period of 8 hours and equal volume of fresh dissolution medium was used to maintain a constant volume. The aliquot samples were filtered and the drug concentration was determined by ultraviolet (UV) method at 293 and 271nm for DAP and CLXS respectively. The cumulative amount of drug released from ethosomes was plotted against time [17]. To know the mechanism of drug release from these formulations, the data were treated according to first order (log cumulative percentage of drug remaining versus time), Higuchi’s (Cumulative percentage of drug released versus time) and Korsmeyer’s (log cumulative percentage of drug released versus log time) equation along with a zero-order (Cumulative percentage of drug release versus time) pattern.
2.10. Stability Studies
The purpose of the stability study is to provide evidence of the quality of a drug substance or drug product which varies with time under the influence of a verity of environmental factors such temperature, humidity and light. A stability study was performed on randomly selected three ethosomal formulations, in which phospholipid, cholesterol and propylene glycol percentage were varied. Ethosomes were stored under static conditions 4°C, (room temperature) 25°C and at high temperature 60°C in glass over a periods of one month. Results were expressed in terms of Drug content [18].
2.11. Preparation of Gel with Optimised Ethosome
A numerical optimization technique using desirability approach was employed to select an optimized formulation in the desired responses. Constraints like Zeta potential, maximizing entrapment efficiency and % drug release at the end of 8 hours as well as minimizing the particle size were set as a goal to select the optimized formulation. Gels loaded with composite ethosomal vesicles were prepared by dispersion method. Carbopol 934 was soaked in distilled water (1%w/v) for 2 h to form its aqueous dispersion. To this dispersion, permeation enhancer polyethylene glycol 400 and isopropyl myristate and combinations thereof were added as per the composition (Table 2). Optimized ethosomal formulation was centrifuged at 2000 rpm and vesicles were incorporated in Carbopol gel base to get G5. Triethanolamine was added under continuous stirring to the above dispersion. Formulations G1-G3 were designated as plain gels whereas ethosomal optimized gel is G5 [18]. Preparation of gel was done manually by accurately weighing the optimized ethosome which contains the equivalent amount of 75mg/g DAP and CLXS.
|
Ingredients %w/w |
Formulation code | ||||
|---|---|---|---|---|---|
| G1* | G2** | G3$ | G4# | G5@ | |
| Drug (DAP, CLXS) | 75 | 75 | 75+75 | - | Optimised Ethosome |
| Optimized ethosome | - | - | - | - | - |
| Carbopol 934 | 1 | 1 | 1 | - | 1 |
| PEG 400 | 5 | 5 | 5 | - | 5 |
| Isopropyl myristate | 5 | 5 | 5 | - | 5 |
| Triethenolamine | 2 | 2 | 2 | - | 2 |
| Water | 100 | 100 | 100 | - | 100 |
2.12. Evaluation of Prepared Gel
2.12.1. Determination of Gel Viscosity
The viscosity of the formulations and reference was performed using a Brookfield digital viscometer (Model DV-II, USA) equipped with spindle S27. The apparent viscosity was measured at 17 sec−1 shear rate (50 rpm) and room temperature, after a 3 min rest time [19].
2.12.2. Determination of Gel pH
One gram each of the gel formulations and the reference was accurately weighed and dispersed in 10 ml of purified water. The PH of the dispersions was measured with a PH meter (Hanna Instruments, HI8417, Portugal) [20].
2.12.3. Compressibility or Spreadability
24 hr old gels was placed between two horizontal plates of 20 cm2, of which the upper one weighted 46.36 gm and 200gm weight was placed over it at ambient temperature. A circle of 5 mm in diameter was made and the diameter of gel was measured after 5 minutes [21].
2.12.4. Skin Irritation Study
Skin irritation study was carried out on healthy rats (150-200 g) (IAEC/PSIT/1273/ac/15). The animals were maintained on standard animal feed and had free access to water. Before one day of starting the study hair was shaved from back and area of 5 cm2 was marked on both the sides, one side served as control while the other side was test. Prepared ethosomal gel was applied for 7 days and the site was observed for any sensitivity and the reaction if any and graded as 0, 1, 2, 3 for no reaction, slight patchy erythema and slight but cofluent or moderate but patchy erythema respectively [20, 22].
2.13. Determination and Evaluation of Drug Release Studies
Drug release study was performed by using Franz diffusion cell and the release kinetic was studied by various kinetic models as zero order plot, first order plot, higuchi plot and korsmeyer-peppas plot. To study the release kinetics of the ethosomal gel data obtained from in-vitro drug release studies were plotted in various kinetic models: zero order as the cumulative amount of drug releases Vs. Time, First order as log cumulative % of drug remaining Vs. Time, higuchi model as cumulative % of the drug released Vs. Square root of time and korsmeyer-peppas model as log cumulative % drug release Vs. Log time [17].
3. RESULTS AND DISCUSSION
3.1. Phase Solubility and in vitro Adsorption Studies
Phase solubility study was performed to optimize the level of cholesterol for the experimental design. Results revealed that solubility has been increased with increase in concentration of cholesterol, but within a particular limit upto 0.6%w/v (Fig. 3). Thus, 0.6% w/v cholesterol concentration has been optimized for ethosomal preparation.
With the aim to better understand physical events occurring in pharmaceutical preparations containing soy lecithin it is important to investigate surface potential of lecithin dispersions in water. Lecithin does not dissolve completely in water. Adsorption study has been done for 24 hrs and it was observed that initially adsorption of drug on soy lecithin was greater due to presence of more activity sites on its surface, but after 1 hr adsorption became gradually decreased, due to abatement of adsorption site on surface of soy lecithin (Fig. 3).
 |
Fig. (3). DAP and CLXS (A) Phase solublity studies; (B) In Vitro Adsorption Studies. |
3.2. Evaluation of Prepared Ethosomes
To identify the presence of organic functional groups in the prepared formulations FT-IR study was performed. In the spectrum of DAP, the strong stretching vibration of the sulfone group O=S=O was at 1108 cm-1 and the bending vibration of p-disubstituted aromatic ring at 831 cm-1. Peaks at 1598 cm-1 and 3367 cm-1 correspond to the N-H bending vibration and stretching vibration respectively. Similar peaks of DAP were found in the spectra of F1 to F9 formulation. In the spectrum of CLXS-C=O peak shown at 1589 cm-1, Aromatic ring peak at 780 cm-1 and N-H peak at 3400 cm-1. All vibrations respectively with the similar peaks of CLXS were found in the spectra of F1 to F9 formulation, which revealed that there was not any type of chemical reaction between drug-drug and drug-excipients. The spectra of phospholipids showed aliphatic C-H bands from 2863.69 cm-1 to 2922.86 cm-1, C=O bands at 17,400 cm-1 and C=C stretching bands from 1588.28 cm-1 to 1519.92 cm-1. Similar peaks were found in the spectra of F1 to F9 formulations (Fig. 4).
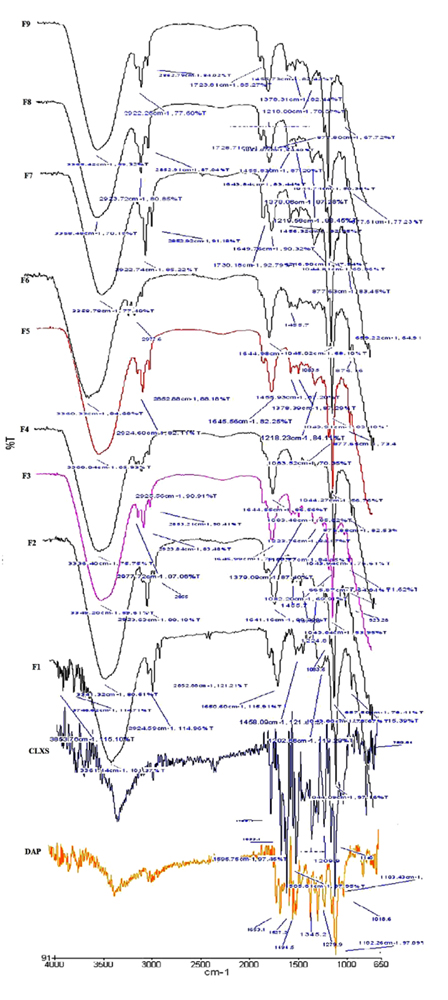 |
Fig. (4). FTIR spectra of Ethosomes prepared with DAP and CLXS. |
Ethosome obtained were spherical shaped, which could have an impact on drug-release. According to SEM images, ethosome ranged in size from 127 to 215 nm. In an ideal condition ethosomes have an average size of 100 to 120 nm. An increase in ethanol concentration (from 36% to 40% ethanol) generally decreases the vesicle size of ethosome. However, a high concentration of ethanol leads to the interdigitation of the lipid bilayers and destabilization of the vesicles. No drug crystals were visible in SEM images (Fig. 5a and 5b), regardless of the preparation technique or the loaded drug. Largest vesicles were observed in formulation F8 containing 36% v/v ethanol (215±7.23 nm) and smallest in formulation F1 containing 40% v/v ethanol (127±9.01 nm). Ethanol significantly reduces vesicle membrane thickness, probably as a result of interpenetration of hydrocarbon chain (ethanol) in the vesicular lipid bilayers. Furthermore, ethanol imparts a net negative charge to the ethosomal system and confers it some degree of steric stabilization that may finally lead to a reduction in vesicular size. The smooth surface of vesicles was confirmed by SEM.
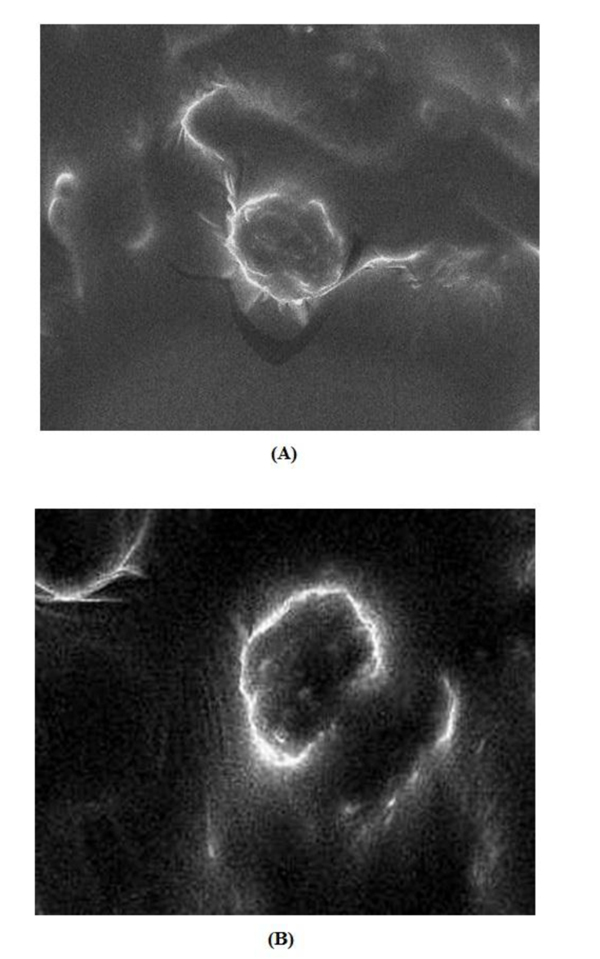 |
Fig. (5). SEM images of ethosomal formulations (A) F2, (B) F1. |
A high zeta potential (> -30 mV) is beneficial to vesicles physical stability as it prevents aggregation between vesicles owing to electrostatic repulsion. If all the vesicles have a large negative or positive zeta potential, then they will tend to repel each other and there will be no tendency for the vesicles to come together. However, if the vesicles have low zeta potential values, then there will be no force to prevent the vesicles to come together and flocculate. The high ethanol concentration in ethosomes has shifted the vesicular charge from positive to negative. It was observed that F1 and F2 ethosomes were having zeta potential of -48.01 mV and -50.11 mV respectively and do not aggregate rapidly. The charge of vesicles is an important parameter that can influence both stability and skin vesicle interaction. From the above results, it was clear that both the presence of ethanol and drug were decisive factors, which determine the surface charge of the ethosomal vesicles. A pareto chart was generated which illustrated that the formulations on the left side of the intersection cause vital effect and formulations on the right hand side has trivial effect on dependent variables. Considering the same, it was observed in Pareto analysis that F2, F1, F5, F9, F6, F4 and F8, F4, F5, F7, F6, F9 (Vital few) had positive effect on zeta potential and vesicle size respectively. Results revealed that 80% of effects (Zeta potential, Vesicle size) comes from 20% of causes (Formulations on left hand side of intersection). By Pareto analysis important causes can be identified and further can be applied to optimize formulations. As shown in Fig. (6A and B) F1 and F2 formulations showed better effects with respect of zeta potential (Right hand side of pareto chart) and vesicle size (Left hand side of pareto chart) because small vesicle size and higher zeta potential are essential parameters for an ethosome to be stable.
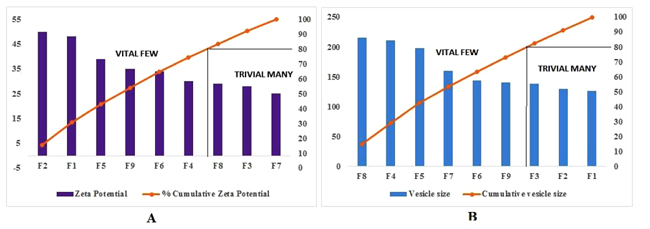 |
Fig. (6). Pareto chart for cause and effect analysis. |
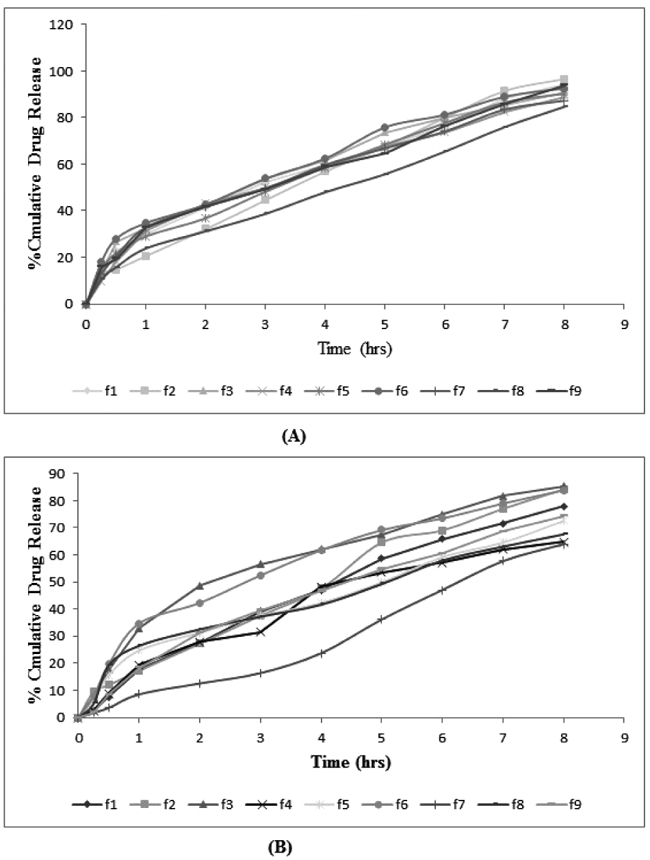 |
Fig. (7). Comparative percentage drug release of (A) DAP and (B) CLXS. |
Entrapment efficiency of the formulations was observed. Ethosomal formulations fabricated with 3.5% phospholipid and 36% alcohol (F5) exhibited 52.31% and 49.07% entrapment efficiency of DAP and CLXS respectively, which was increased to 73.51% and 71.91% when the concentration of phospholipid and alcohol was increased to 4.4% and 40% (F2) respectively but not beyond this level. Higher entrapment efficiency with increased amount of ethanol is possibly due to increased solubility of drug in ethanol present in the ethosomes. Increases in the concentration of permeation enhancer (PG) from 5 to 10% lead to decrease in entrapment due to less flexibility of membrane. PG from 3-5% show increase in entrapment due to its upsurge in flexibility of membrane. Additional rise in concentration of PG probably makes the vesicles more leaky in nature and decrease the entrapment efficiency [23, 24].
3.3. In Vitro Drug Release Study
In Vitro drug release studies of drug loaded ethosomes were conducted for a period of 8 h using Franz diffusion cell, entire formulations were performed on skin pH to evaluate the release. The release profiles of ethosomal formulations were shown in Fig. (7). From the release profile, it was observed that the phospholipid, cholesterol, ethanol and propylene glycol concentration effect drug release profile, these being the main parameters that influenced size and encapsulation efficiencies.
A significant increase in amount permeated from F1 and F2 ethosomal systems was observed as compared to other formulations which could be due to sustained release behavior of system. This may be due to concentration of propylene glycol and its ability to decrease the diffusional resistances of the membrane barrier, possibly because drug kept in dissolved form in the glycol, and diffuses through the membrane. There was an increase in the amount of drug release with the gradual increase in concentration of ethanol from 30 to 40% v/v. This may be due to the effect of ethanol, which was also acting as a penetration enhancer and is responsible for reducing vesicle size. It plays an important role in ethosomal systems by giving the vesicles unique characteristics in terms of size, ζ-potential, stability, entrapment efficacy, and enhanced skin permeability. Kinetic analysis of the in vitro release data and the mechanism of drug release were estimated after fitting the obtained data to different kinetic models; zero-order, first-order, Higuchi, Korsmeyer-Peppas and hixcon crowell models (Table 3). The higher correlation coefficient was judged to be more appropriate model for dissolution data. F1 formulation obey zero order release which is ideal for transdermal drug delivery as it provide constant release of drug over an extended period of time and reflect improvement in therapeutic index.
|
Formulation code |
Zero Order r2 |
First order r2 |
Higuchi r2 |
Korsmeyer-peppas | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| - | DAP | CLXS | DAP | CLXS | DAP | CLXS | DAP | CLXS | ||||
| r2 | N | Diffusion | r2 | N | Diffusion | |||||||
| F1 | 0.989 | 0.993 | 0.926 | 0.976 | 0.98 | 0.977 | 0.9949 | 0.517 | Non-Fickian diffusion | .9794 | .940 | Non-Fickian diffusion |
| F2 | 0.990 | 0.987 | 0.893 | 0.978 | 0.9635 | 0.966 | 0.9752 | 0.6311 | Non-Fickian diffusion | .9859 | .677 | Non-Fickian diffusion |
| F3 | 0.9702 | 0.9933 | 0.9826 | 0.9781 | 0.9954 | 0.9842 | 0.984 | 0.5163 | Non-Fickian diffusion | .911 | .645 | Non-Fickian diffusion |
| F4 | 0.973 | 0.9784 | 0.9731 | 0.9739 | 0.9929 | 0.9495 | 0.9841 | 0.9841 | Non-Fickian diffusion | .946 | .945 | Non-Fickian diffusion |
| F5 | 0.972 | 0.961 | 0.969 | 0.9895 | 0.987 | 0.9789 | 0.9493 | 0.9893 | Non-Fickian diffusion | .975 | .968 | Non-Fickian diffusion |
| F6 | 0.9911 | 0.9733 | 0.9747 | 0.9915 | 0.9632 | 0.991 | 0.9868 | 0.9868 | Non-Fickian diffusion | .9269 | .613 | Non-Fickian diffusion |
| F7 | 0.986 | 0.999 | 0.9822 | 0.8808 | 0.9835 | 0.869 | 0.9924 | 0.9924 | Non-Fickian diffusion | .9773 | 1.01 | Non-Fickian diffusion |
| F8 | 0.9833 | 0.983 | 0.9488 | 0.973 | 0.9676 | 0.976 | 0.9893 | 0.989 | Non-Fickian diffusion | .949 | .613 | Non-Fickian diffusion |
| F9 | 0.9801 | 0.988 | 0.918 | 0.9037 | 0.9823 | 0.983 | 0.988 | 0.989 | Non-Fickian diffusion | .967 | .954 | Non-Fickian diffusion |
3.4. Stability Study
The appearance of optimized ethosomal formulation showed slight changes in 60°C. The drug content of all formulations at 4°C does not show any significant decrease, followed by the formulations kept at 25°C whereas the formulations kept at 60°C showed a significant loss in the drug content. A study showed that ethosomes were not stable at high temperature and caused loss of drug content (Fig. 8). As a result of stability studies, it can be concluded that the formulations are most stable when stored at a lower temperature i.e. 4°C.
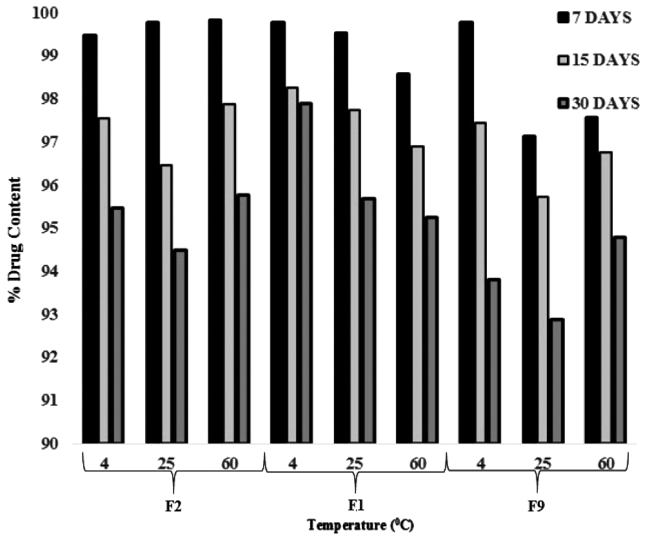 |
Fig. (8). Stability study of DAP and CLXS loaded ethosomes at different temperatures for 7, 15 and 30 days. |
3.5. Preparation and Evaluation of Ethosomal Gel
Ethosomal gel was prepared by dispersion method. Prepared ethosomal gel was compared with marketed formulations. Gels were transparent with faint odor of raw materials used. The pH of ethosomal gels were evaluated and the value was found to be in range of 6.8 ± 0.35 and 7.4 ± 0.45 (Table 4). pH of all formulations lies between the normal physiological pH range of the skin, i.e., 3.0-9.0 [25], which justify non-irritating effect of ethosomal gel on skin. Viscosity of all the gels was found in the range of 42158 ± 1.47 and 46115 ± 1.11 cp. Determination of viscosity of gel matrix assists in evaluating the penetration across the skin by controlling the release of the drug into receptor medium. The gel was homogeneous in nature and there was no any type of coarse particles was appeared after visual appearance. All ethosomal gel formulations were applied for 7 days on healthy rats and the site was observed for any sensitivity and reaction if any. After 7 days skin irritation was observed and scores have been given on the basis of irritation, slight and moderate patchy erythema from ethosomal gel. It was observed that there was no irritation after application of ethosomal gels. Spreadability studies were determined by using the two horizontal plates and the results were compiled in Table 4. Formulation G4 showed highest spreadability compare to all other formulations and thus will give better patient compliance.
| S.No. | Formulation code | Viscosity (centipoise) | pH |
Sensitivity Grade* |
Spreadability |
|---|---|---|---|---|---|
| 1 | G1 | 46115±1.11 | 7.0 | 0 | 11.05 |
| 2 | G2 | 45223±0.92 | 7.4 | 2 | 10.05 |
| 3 | G3 | 47889±1.13 | 7.0 | 1 | 11.78 |
| 4 | G4 | 45778±0.11 | 6.8 | 0 | 12.68 |
| 5 | G5 | 42158±1.47 | 6.9 | 0 | 12.05 |
In vitro skin permeability data of DAP and CLXS from the formulated gels was obtained. Highest and sustained in vitro permeation was found in gels containing ethosomal vesicles (G5) (Fig. 9). G5 obey zero order release is ideal for transdermal drug delivery as they provide constant release of drug over an extended period of time and reflect improvement in therapeutic index. On the basis of results obtained, G5 was selected as optimized gel among other gels as it was able to reach the target flux and showed greater percent cumulative drug permeability significantly different to other gels (p>0.05). This may be due to its higher flexibility and the ability to retain vesicle integrity [25]. Permeability enhancement was observed due to synergistic effect of propylene glycol and ethanol [26]. Propylene glycol acts as a penetration enhancer for the drugs which are better soluble in alcohol than in water. The solvation of keratin within the stratum corneum by competition with water for the hydrogen bond binding sites and the intercalation in the polar head groups of the lipid bilayers by propylene glycol are postulated as mechanisms of action for the penetration enhancement [27]. The synergistic combinatorial effect of ethanol and soya lecithin is depicted to be responsible for a deeper drug loaded distribution and penetration into the skin layers [28].
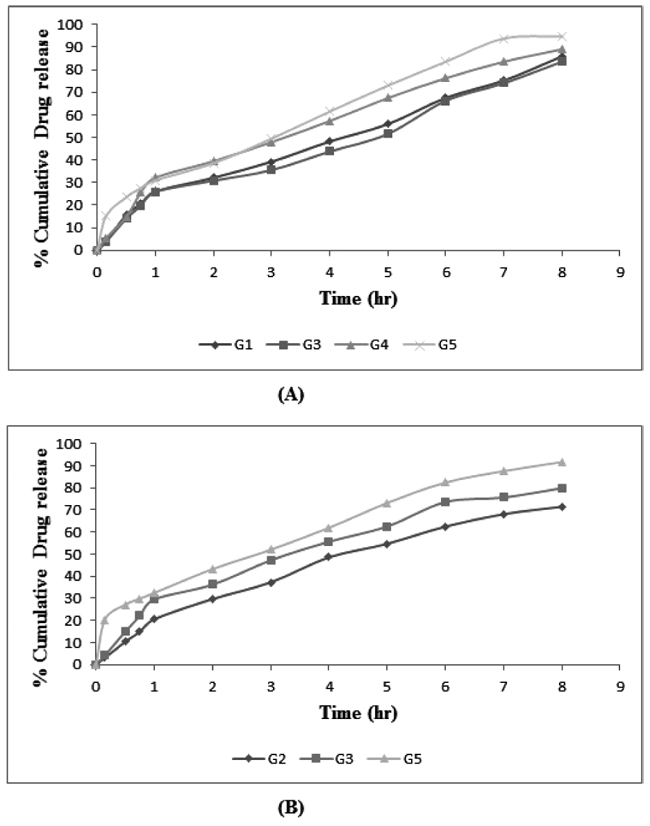 |
Fig. (9). In vitro permeability profile of (A) DAP and (B) CLXS from different gel formulations in phosphate buffer pH 6.8. |
CONCLUSION
Ethosomal carriers in gel matrix represented a stable and efficient topical delivery of DAP and CLXS with higher entrapment efficiency, and stability than reference ethosomal vesicles. In vitro permeation study confirmed the uniform and sustained permeation of drugs via ethosomal gel. In case of Leprosy, patients take lots of Antileprotic drugs & Antibiotics orally for the treatment of the disease. So Ethosomal gel of DAP and CLX can be the best treatment option without any side effect. Ethosomal gel not only offers a promising means of drug delivery, but also could enhance the recovery rate of the skin barrier.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The experimental protocol was approved (IAEC/PSIT/1273/ac/2017) by the Institutional Animal Ethical Committee prior to the conduct of the animal experiments. Study has been conducted on preclinical basis.
HUMAN AND ANIMAL RIGHTS
No humans were involved in the study. The reported experiments were in accordance with the standards set forth in the 8th Edition of Guide for the Care and Use of Laboratory Animals (http://grants.nih.gov/grants/olaw/Guide-for -the-care-and-use-of-laboratory-animals.pdf) published by the National Academy of Sciences, The National Academies Press, Washington DC, United States of America.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLDEGEMENTS
Declared none.
